To model large-scale systems and the effects of complex environments, we will develop multiscale tools coupling quantum/atomistic models to coarse-grain/mesoscale schemes, spanning several orders of magnitude in space and time.
New developments in experimental techniques allow complex phenomena and processes to be resolved in both space and time. The dimensionality of the processes that can be investigated span several orders of magnitude, from fast highly localized chemical reactions occurring on the picosecond timescale and localized to a few nanometers, to large-scale diffusional processes extending beyond micrometers and milliseconds. Although all systems obey the same fundamental principles of quantum mechanics, phenomena at different resolutions do not require descriptions at the same level of detail [1]: Whereas spectroscopic properties require a quantum-mechanical description, molecular localization may be described even without atomistic resolution. Multiscale modelling ensures a balanced treatment of phenomena at different scales, see the figure below [2].
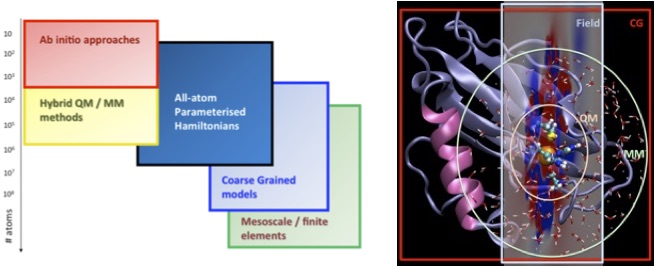
In RT2, detailed representations based on electronic-structure methods, including those developed in RT1, will be embedded in a multiscale hierarchy of progressively coarser descriptions of the environment: QM/QM, QM/MM, QM/PCM [3, 4]. These models will give accurate descriptions of the dielectric response of the environment, taking also into account steric effects, thus providing crucial enabling tools for the research in RT3–RT5, while also contributing to RT6. The development of embedding schemes for multidimensional vibrational spectroscopy has recently received RCN-RP support (Ruud) and will be vigorously pursued in RT2.
At the other end of the scale, we will work on multi-resolution atomistic/coarse-grained/mesoscale simulations, developing, in particular, the hybrid particle–field molecular-dynamics self-consistent-field (MD-SCF) method [5]. This method describes particle–particle interactions by coupling the individual particles to an external potential dependent on the density. In this manner, intermolecular forces can be calculated in linear time, allowing simulations of quasi-atomistic resolution at scales that are today hardly accessible even on the most powerful massively parallel architectures. In RT2, we will extend the MD-SCF method to biological systems and biomaterial interfaces. Seed funding has been obtained from the H2020 Marie Skłodowska-Curie Action (MSCA) by Bindu and Cascella. All multiscale and multiresolution methods will be integrated in codes developed at the centre; in col- laboration with Prof. G. Milano, our MD-SCF developments will also be incorporated in the OCCAM code. In the long term, we seek a unifying description connecting all resolutions up to the mesoscale.
[1] M. L. Klein et al. “Large-scale molecular dynamics simulations of self-assembling systems”. Science 321 (2008), 798. [2] M. Cascella et al. “Toward accurate coarse-graining approaches for protein and membrane simulations”. Specialist Periodic Report – Chemical Modelling. Vol. 12. Royal Society of Chemistry, 2015, p. 1. [3] E. Brunk et al. “Mixed Quantum Mechanical/Molecular Mechanical Molecular Dynamics Simulations of Biological Systems in Ground and Electronically Excited States”. Chem. Rev. 115 (2015), 6217. [4] J. Tomasi et al. “Quantum Mechanical Continuum Solvation Models”. Chem. Rev. 105 (2005), 2999. [5] G. Milano et al. “A hybrid particle-field molecular dynamics approach: a route toward efficient coarse-grained models for biomembranes”. Phys. Biol. 10 (2013), 045007.