Proteomics
The proteome is defined as all the proteins related to a genome; in fact, due to processes such as alternative splicing and post-translational modifications, the proteome is even more complex that its corresponding genome. Proteomics is the study of the proteome and aims to describe and characterise all of the expressed proteins in a biological system.
Apoptosis
Apoptosis is a tightly regulated form of programmed cell death (PCD) and is important for removing unwanted cells without causing inflammation. In fact, defects in apoptotic signaling have been shown to be of central importance in the pathogenesis of most human diseases. Therefore, apoptosis modulation offers exciting therapeutic opportunities for the treatment of such diseases.
First, however, we must understand the proteins involved in apoptosis and their specific roles in diseases in order to evaluate potential therapies. The processes involved in apoptosis are complex and include the regulated proteolytic cleavage of specific target proteins by caspases, posttranslational modifications, protein-protein interactions, and protein translocations. Proteome analyses of the different signaling pathways of apoptosis can be used to identify the specific proteins that are modified during the process.
Chemotherapy
Chemotherapeutic drugs often kill cancer cells by inducing apoptosis in rapidly dividing cells. Unfortunately, there are many complications with chemotherapy, including unwanted side effects and the development of drug resistance.
A better understanding of the molecular mechanism of these drugs would help facilitate the optimization of chemotherapy. Proteome profiling, the comparison of drug-treated apoptotic cells to untreated cells, provides a set of target protein markers. These identified proteins are then analysed in detail, as the signals to activate or prevent apoptosis commonly depend on the post-translational modification of apoptosis regulators (e.g., cleavage by caspases, phosphorylation and translocation between different cellular compartments).
Prostate cancer
Prostate cancer (PCa) is the most common male malignancy in the Western world and a leading cause of cancer death. Elevated prostate-specific antigen (PSA), abnormal digital rectal examination (DRE) and transrectal ultrasonography (TRUS) are the principle methods assessing prostate cancer. In current practice, serum PSA levels of ≥ 3 - 4 ng/ml are typically used to predict the presence of prostate cancer. However, several studies indicate that many high-risk cancers are also detectable in patients with lower PSA levels. Furthermore, benign condition can also cause PSA levels to increase and thus PSA is not necessarily very specific to prostate cancer. Moreover, large screening studies revealed mixed conclusions about the benefit of PSA screening. Actually, overdiagnosis and overtreatment of asymptomatic and harmless prostate cancers were also performed after testing PSA.
More accurate biomarkers to diagnose prostate cancer with minimally invasive methods are highly demanded to reduce the number of missed cases, to avoid biopsies of patients with benign pathologies, and to follow up either active surveillance as well as postprostatectomy. A test in serum, plasma or urine would further reduce costs and patient morbidity. In particular, large collections of serum, plasma and urine in biobanks are available in many countries and can be utilized to find biomarkers or even better panels of biomarkers for a more precise prediction of diseases. A panel of a few proteins including PSA can certainly increase the specificity and sensitivity to detect prostate cancer.
Therapeutic proteins
Therapeutic proteins are attractive anti-cancer agents due to their high target specificity and minimal toxicity in comparison to small-molecule drugs. Most therapeutic proteins are produced by recombinant DNA technology and purified from a wide range of organism. However, the production of therapeutic proteins is associated with the generation of a mixture of very similar protein forms, so-called protein species or proteoforms. Some of them are poorly effective or even harmful. Therefore, protein analytical tools are required to guarantee the qualitative and quantitative detection of the diverse protein species.
Proteomics platform at IBV/UiO
The proteomics group uses a variety of techniques in their examination of apoptosis signalling. An overall view of the proteomics platform is shown in the figure below, followed by brief descriptions of the techniques involved.
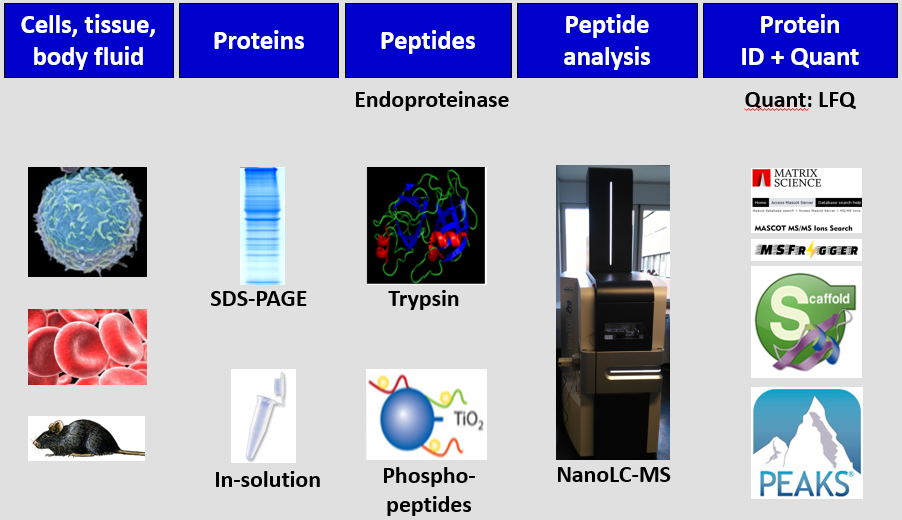
- Model organism - human tumour cell lines and body fluids.
- Induction of apoptosis - chemotherapeutic drugs: Cisplatin - causes DNA damage; Taxol and STLC - cause mitotic arrest; Sorafenib - triggers ER stress-induced apoptosis.
- Protein separations - liquid chromatography; gel electrophoresis.
- Protein identification - by mass spectrometry:
- ESI mass spectrometry - used to identify complex protein samples, usually by direct coupling to nano-liquid chromatography, by sequencing with tandem-MS (MS/MS). Currently, we have a TimsTOF Pro system.
- Quantitative protein analysis:
- LFQ (Label-free quantification) - no labeling required.
- Posttranslational modifications: co-immunoprecipitation; affinity chromatography; TiO2 beads; phosphoproteomics, glyco analysis.
- Protein-protein interactions: co-immunoprecipitation. This technique is used in combination with proteomics approaches.
- Bioinformatics - several bioinformatics tools are used for handling and analysing the comprehensive data: Mascot; MSFragger; Scaffold; MaxQuant; and PEAKS.
- Proteomics databases: consolidation of published proteomics data (cancerproteomics.uio.no).
- IsobariQ - software designed by Thiede Group for relative quantification of IPTL, iTRAQ or TMT labelled peptides (download software).
Proteomics Service
The Thiede group also runs the UiO Proteomics Core Facility, with services available for both internal and external customers.